One fundamental problem that persists in this analysis is the efficient alignment of in-silico digested contigs to an optical map. We develop Twin to tackle this very problem. Twin is the first index-based method for aligning in-silico digested contigs to an optical map. Our results demonstrate that Twin is orders of magnitude faster than competing methods on a range of genomes.

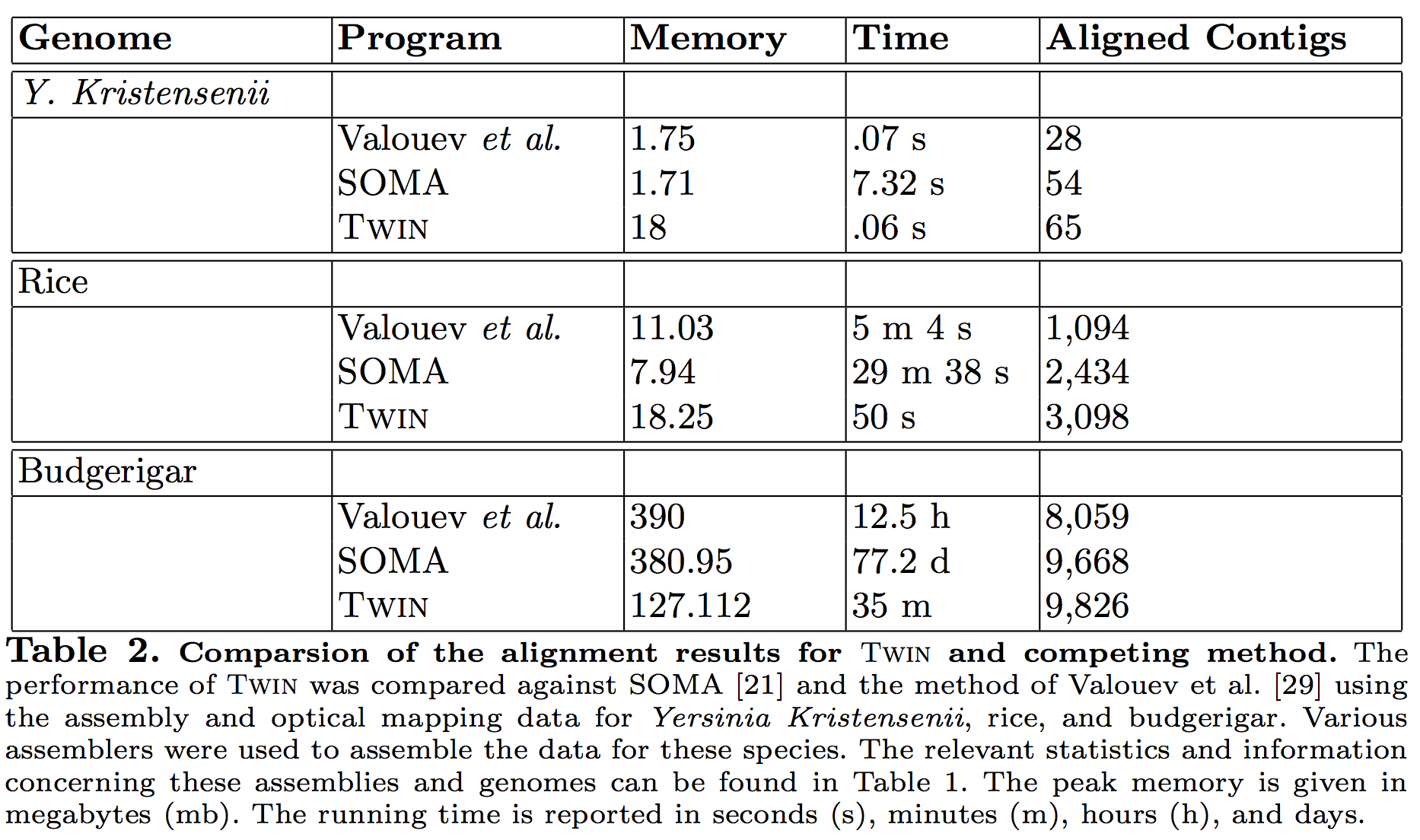
Most-importantly, it is specifically designed to be capable of dealing with very large eukaryote genomes and thus, is the only non-proprietary method capable of completing the alignment for the budgerigar genome in a reasonable amount of time. The genome assemblies, optical maps, and files detailing the reads and assembly process are available for download at gigadb.org/dataset. Table 1 demonstrates the statistics on the assemblies generated using short read data and Table 2 illustrates the performance of Twin with respect to that of competing tools on the assemblies of these genomes and their accompanied optical map. ß
Funding was provided by the National Institutes of Health through the Colorado Clinical and Translational Sciences Institute (CCTSI).