
Projects¶
The main focus of research in my lab is on the development of machine learning methods for problems in bioinformatics. Our lab pioneered the use of graph convolutional networks for the analysis of protein structures. We are also applying deep learning techniques for extracting signals from genomic sequences.
Deep learning in genomics¶
Genes, that contain the information that codes for mRNA and proteins, are constantly turned on and off in response to the needs of the cell and the organism that they are a part of. The molecular switches that control genes are proteins that bind DNA. These DNA binding proteins (transcription factors) recognize signals in the DNA. The complexity of the genome and the molecular processes that controls its expression are one of the major challenges facing biologists. We are using deep learning techniques to help discover these signals and help generate models that can help unravel various biological processes (see the discussion of alternative splicing below). Here are some examples of recent work in this area:
We designed a deep learning algorithm that allows for the discovery of interactions between regulatory features by leveraging self-attention:
- Fahad Ullah and Asa Ben-Hur. A self-attention model for inferring cooperativity between regulatory features. Nucleic Acids Research, 2021.
We used deep learning to understand the process of alternative splicing:
- Fahad Ullah, Saira Jabeen, Maayan Salton, ASN Reddy, and Asa Ben-Hur. Evidence for the role of transcription factors in the co-transcriptional regulation of intron retention Genome Biology 24, 53, 2023.
A follow up paper posits that large scale chromatin models are the foundation models of genomics:
- Ahmed Daoud and Asa Ben Hur. Using large scale transfer learning to highlight the role of chromatin state in intron retention. bioRxiv 2024.01.26.577402, 2024.
Our earlier work is a comparative study of deep learning architectures for discovering signals in DNA and RNA:
- Ameni Trabelsi, Mohamed Chaabane, and Asa Ben-Hur. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics, 35:14, i269–i277, 2019 (ISMB 2019 special issue). (preprint)
Analyzing protein structures with graph neural networks¶
My lab has been studying protein-protein interactions and other aspects of protein function. Proteins perform their function by interacting with other proteins. Therefore understanding the complex network of interactions between proteins, and at a finer level, determining the interfaces through which those interactions occur are highly important. To address the problem of predicting interfaces, we have introduced the concept of graph convolution to the analysis of protein 3d structures.
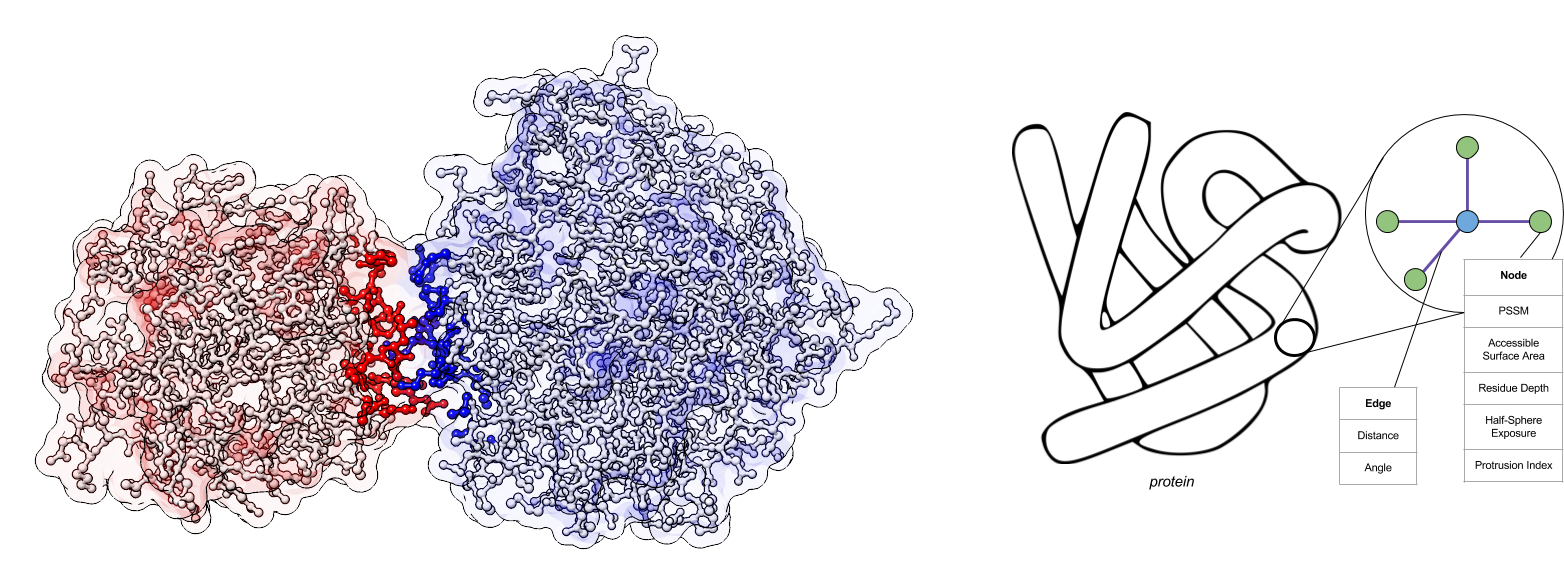
This was inspired by the success of convolutional networks in computer vision. Their power comes from their ability to learn features with increasing levels of abstraction that are invariant to various transformations of the image (e.g. translation and rotation). We envisioned a similar approach for protein 3d structures. However, instead of representing proteins as a 3d image, we chose to represent the structure as a graph whose nodes represent atoms or amino acids, with connections that are determined by proximity in the protein structure. This required us to replace the standard form of convolution, which operates over a regular grid, to convolution over a graph structure. Our first results using this technique were published at the NIPS conference:
- Alex Fout, Jonathon Byrd, Basir Shariat, and Asa Ben-Hur. Protein interface prediction using graph convolutional networks. In: Advances in Neural Information Processing Systems (NIPS), 2017. (code, data, poster)

We have recently applied this approach to the problem of assessing the quality of predicted protein structures, introducing a novel loss function inspired by the SVM regression epsilon-insensitive loss.
- Soumyadip Roy and Asa Ben-Hur. Protein quality assessment with a loss function designed for high-quality decoys Frontiers in Bioinformatics 3, 2023.
This project was funded by a grant from the NSF ABI program (award # 1564840).
Our proof-of-concept for the feasibility of partner specific prediction of interfaces from 3d structure used SVMs and resulted in a method called PAIRpred:
- Fayyaz Afsar Minhas, Brian Geiss, and Asa Ben-Hur. PAIRpred: Partner-specific prediction of interacting residues from sequence and structure, PROTEINS: Structure, Function, and Bioinformatics, 2013. PAIRpred software and data.
Earlier work in the area of interface and interaction prediction includes prediction of Calmodulin binding sites, and genome-wide prediction of interaction networks in yeast and human:
- Fayyaz Afsar Minhas and Asa Ben-Hur. Multiple instance learning of Calmodulin binding sites. Bioinformatics (2012) 28(18): i416-i422 (special ECCB 2012 issue). Here’s a link to a webserver with an implementation of the method.
- H. Wang, E. Segal, A. Ben-Hur, Q. Li, M. Vidal and D. Koller. InSite: a computational method for identifying protein-protein interaction binding sites on a proteome-wide scale. Genome Biology, 8(9): R192, 2007.
- Asa Ben-Hur and William Stafford Noble. Kernel methods for predicting protein-protein interactions. In: Proceedings, thirteenth international conference on intelligent systems for molecular biology. Bioinformatics 21(Suppl. 1): i38-i46, 2005.
Deep learning tools for basecalling nanopore RNA sequencing data¶
Oxford Nanopore Technology sequencing devices are capable of directly sequencing long read RNA as is, with the potential of being able to detect modified bases without needing special sample preparation. However, todate there is no such tool available, that would enable easy access to the epitranscriptome.
In a preliminary study towards this goal we concentrated on improving RNA basecalling accuracy. We designed a novel basecalling architecture achieving state-of-the-art performance, improving on the accuracy of the commercial basecaller from Oxford Nanopore Technologies. This basecaller, called RODAN is freely available on github.
- Don Neumann, ASN Reddy, and Asa Ben-Hur. RODAN: a fully convolutional architecture for basecalling nanopore RNA sequencing data. BMC Bioinformatics 23, 142, 2022.
We are currently completing work on the detection of post-transcriptional RNA modifications in nanopore sequencing data with a focus on the detection of methylated adenosines, known as m6a . Our neural network, whose codename is Mothra, is the first of its kind, as it is capable of simultaneously basecalling and discerning modifications with read level resolution. Its architecture is based on the architecture developed in RODAN, and uses attention layers to pinpoint modified bases within a sequencing read. Our work will facilitate research into the detection of m6a while also furthering progress in the detection of other post-transcriptional modifications.
This project was funded by an NSF EAGER grant (award # 1949036).
Alternative splicing¶
Splicing is the process whereby parts of a gene called introns are removed, and the RNA is spliced back to form the mature mRNA. A given gene can be spliced in multiple ways, a phenomenon called alternative splicing. Whereas it is well-studied in animals, alternative splicing in plants is not as well understood, and the differences in genome architecture between plants and animals lead to differences in alternative splicing. We are working on this in collaboration with A.S.N. Reddy of the Biology Department. Our approach is to computationally search for genomic features that are predictive of alternative splicing—elements that serve as splicing enhancers and suppressors, and test their biological relevance to the process.

The above figure shows a model created by our SpliceGrapher tool.
This project was funded by NSF and DOE.
- Salah E. Abdel-Ghany*, Michael Hamilton*, Jennifer L. Jacobi, Peter Ngam, Nicholas Devitt, Faye Schilkey, Asa Ben-Hur, and A.S.N. Reddy. A survey of the sorghum transcriptome using single-molecule long reads. Nature Communications 7, 2016. (*Joint first authors). Here’s a link to the TAPIS software.
- M.F. Rogers, J. Thomas, A.S.N. Reddy, and A. Ben-Hur. SpliceGrapher: Detecting patterns of alternative splicing from RNA-seq data in the context of gene models and EST data. Genome Biology 13:R4, 2012.
Protein function prediction¶

Protein function prediction is an ongoing area of research in the lab. The difficulty in applying state-of-the-art machine learning methods to this problem is that proteins can have multiple functions, and that the system of keywords used to describe protein function, the Gene Ontology (GO), has a complex hierarchical structure. This provides genome annotators with a rich vocabulary with which to describe protein function, but makes it sub-optimal to use standard machine learning approaches. We addressed protein function prediction as a hierarchical multi-label classification problem and designed custom algorithms based on the so-called structured SVM, which is able to fully model the complexity of this learning problem. The method we developed, GOstruct, has shown state-of-the-art performance in several benchmarks.
This project was funded by an NSF grant from the ABI program (award #0965768).
- Automated function prediction competition participants. A large-scale evaluation of computational protein function prediction. Nature Methods, 10:221–227, 2013.
- Artem Sokolov, Chris Funk, Kiley Graim, Karin Verspoor, and Asa Ben-Hur. Combining heterogeneous data sources for accurate functional annotation of proteins. Automated Function Prediction Meeting Proceedings (ISMB 2011). BMC Bioinformatics, 14(Suppl 3), 2013.
- A. Sokolov and A. Ben-Hur. Multi-view prediction of protein function. In: ACM Conference on Bioinformatics, Computational Biology and Biomedicine, 2011. [ preprint ]
- A. Sokolov and A. Ben-Hur. Hierarchical classification of Gene Ontology terms using the GOstruct method. Journal of Bioinformatics and Computational Biology 8(2): 357-376, 2010. [ preprint ]
- M. Rogers and A. Ben-Hur. The use of Gene Ontology evidence codes in preventing classifier assessment bias. Bioinformatics 25(9):1173-1177, 2009. [ preprint ]